> For the complete documentation index, see [llms.txt](https://docs.labii.com/llms.txt). Markdown versions of documentation pages are available by appending `.md` to page URLs; this page is available as [Markdown](https://docs.labii.com/widgets/section-widgets/biology/bioinformatics/crispresso2.md).
# CRISPResso2
## **Specs**
Label
Value
Version
0.1.0 (updated on 2021-09-15)
Developer
Labii Inc.
Type
Section
## Overview
[CRISPResso2](https://github.com/pinellolab/CRISPResso2) is a software framework crafted to facilitate swift and user-friendly analysis of genome editing experiments. You can access a basic web version of it at: .
In essence, CRISPResso2 performs the following tasks:
1. Aligns sequencing reads to a reference sequence.
2. Measures insertions, mutations, and deletions to ascertain whether a read has been altered or remains unmodified by genome editing.
3. Presents the editing outcomes in easily comprehensible plots and datasets.
Labii has developed a CRISPResso2 widget encompassing both a user-friendly frontend interface and backend processing servers. This widget enables the execution of CRISPResso2, utilizing the provided input and parameters. Moreover, it seamlessly integrates the analysis results into your notebook.
## Use case
CRISPResso2 is a versatile tool capable of analyzing genome editing outcomes, whether they involve cleaving nucleases like Cas9 or Cpf1, or noncleaving nucleases such as base editors. It offers an array of automated functions, including:
1. Filtering out low-quality reads.
2. Trimming adapters.
3. Aligning reads to one or multiple reference sequences (in the case of multiple alleles).
4. Quantifying outcomes related to HDR (Homology-Directed Repair) and NHEJ (Non-Homologous End Joining) when the HDR sequence is provided.
5. Quantifying frameshift/inframe mutations and identifying affected splice sites when an exon sequence is provided.
6. Visualizing the distribution and position of indels (insertions or deletions) for cleaving nucleases.
7. Visualizing the distribution and position of substitutions for base editors.
8. Visualizing alleles and their respective frequencies.
Furthermore, CRISPResso can be employed as part of a larger tool suite for more extensive analysis:
* CRISPRessoBatch is used to analyze and compare multiple experimental conditions at the same genomic site.
* CRISPRessoPooled is designed for analyzing multiple amplicons from a pooled amplicon sequencing experiment.
* CRISPRessoWGS is employed to assess specific sites in whole-genome sequencing samples.
* CRISPRessoCompare facilitates the comparison of editing outcomes between two samples, such as treated and control groups.
* CRISPRessoAggregate is a tool for consolidating results from previously conducted CRISPResso analyses.
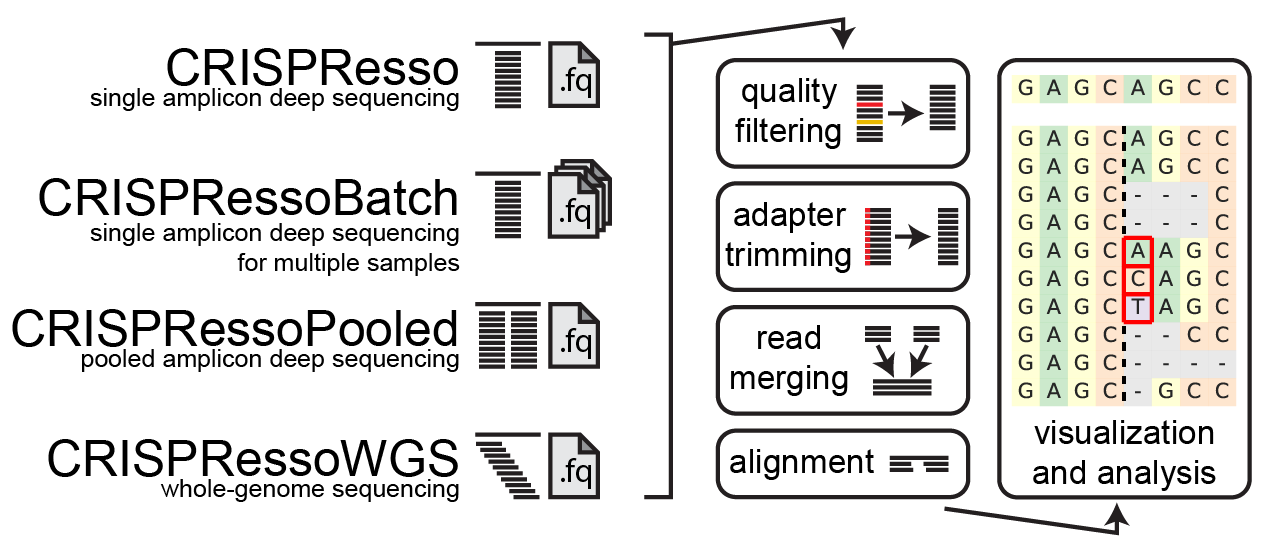
## How does CRISPResso2 work
### **1. Quality filtering**
Input reads are first filtered based on the quality score (phred33) in order to remove potentially false positive indels. The filtering based on the phred33 quality score can be modulated by adjusting the optimal parameters (see additional notes below).
### **2. Adapter trimming**
Next, adapters are trimmed from the reads. If no adapter are present, select 'No Trimming' under the 'Trimming adapter' heading in the optional parameters. If reads contain adapter sequences that need to be trimmed, select the adapters used for trimming under the ‘Trimming adapter’ heading in the optional parameters. Possible adapters include Nextera PE, TruSeq3 PE, TruSeq3 SE, TruSeq2 PE, and TruSeq2 SE. The adapters are trimmed from the reads using Trimmomatic.
### **3. Read merging**
If paired-end reads are provided, reads are merged using FLASh . This produces a single read for alignment to the amplicon sequence, and reduces sequencing errors that may be present at the end of sequencing reads.
### **4. Alignment**
The preprocessed reads are then aligned to the reference sequence with a global sequence alignment algorithm that takes into account our biological knowledge of nuclease function. If multiple alleles are present at the editing site, each allele can be passed to CRISPResso2 and sequenced reads will be assigned to the reference sequence or origin.
### **5. Visualization and analysis**
Finally, after analyzing the aligned reads, a set of informative graphs are generated, allowing for the quantification and visualization of the position and type of outcomes within the amplicon sequence.
## How to use
You can activate the widget by adding it to your record.
### Step 1: Provide Fastq and batch template
The initial step involves supplying the input fastq files along with the corresponding batch template. The fastq files can be uploaded as a folder to AWS S3, while the batch template should be uploaded as a TSV file to the same location.
* **S3 Link:** Provide the AWS S3 link of fastq folder(s). Please make sure the read access has been provided to Labii. For example: s3://xxx/xxx/00\_fastq/.
* **Batch Template:** Please provide the sequence information for the batch template in the specified format. Please note that due to file size limitations, it is not feasible to pass the batch template via the command line. Therefore, it is necessary to upload a batch template file to S3 beforehand. For example: s3://xxx/xxx/xxx.tsv
### Step 2: Analysis and results
After you've provided the fastq and batch template, you can initiate the analysis by clicking on the "**Perform CRISPResso2 analysis**" button. The results will be accessible in an S3 folder.
## References
1. CRISPResso2 source code: